Olympic B3 Science Camps 2016
Bioinformatics Camp -May 2016
May 19th - May 21st
|
|
Vocabulary & Background - References
|
Vocabulary Section: definitions of technical words used in class. Definitions are intended to make class discussions clear and may not be strictly accurate or complete.
Vocabulary Links
Vocabulary List
A:
- Alignment- The arrangement of two or more sequences, of the same type (i.e. amino acid or nucleotide) arranged so that shared parts line up. This makes it easier to find similar and divergent regions. The degree of relatedness (similarity, which may be due to a shared ancestor) between the sequences is assessed by weights assigned to the elements, with the likelihood of differences and similarities estimated compared to some neutral model.
- Alleles - Different sequence forms of a gene that can occupy the same position on the chromosome.
- Amino acid- The monomers, or basic building blocks, of protein polymers. Genes (DNA) code for proteins through the intermediaries of mRNA and tRNA.
- Annotation- labels that describe information about a biological sequence and are connected to that sequence in some way so they can be retrieved. Annotation gives meaning to the sequence: it points to the biologically relevant features in the sequence. Some example are:
- Enzymatic activities of a protein.
- Secondary structure, such as hairpin loops in RNA or alpha-helices in a protein.
- Similarities to other biomolecules.
- Disease(s) associated with mutations in the gene or deficiencie(s) in the gene product.
- Sequence variants and their frequency in a population.
- Assembly - the result of identifying how a collection of short DNA fragments overlap to make a virtual longer fragment. The longer fragment may be called 'the' assembly, or a 'contig' meaning contiguous sequence. Assembly algorithms provide rules for determining how much overlap is required before any two short fragments are considered to overlap, how many times each nucleotide must appear in the collection of sequences, and what the minimal quality of each nucleotide must be before you decide to use it in assembling that sequence, among other things.
B:
- Bases- the core components of the monomer subunits that make up DNA and RNA; the bases share common sugars (ribose for RNA, deoxyribose for DNA making up nucleosides) and phosphate groups (making nucleotides). There are four bases in DNA: adenine, guanine, cytosine, and thymine (A,G,C,T). RNA also has 4 standard bases, replacing the thymine with uracil (U). There are also modified nucleotides, chemically modified versions of the basic 4.
- Base Pairs- The nucleotides are able to form a number of hydrogen bonds between one another - the most famous occurrence is in the DNA double helix, in which two strands of DNA running in opposite directions can form hydrogen bonds that result in a consistent and stable registration between the strands. Classically, in DNA, Adenine pairs with Thymine (or Uracil), and Guanine pairs with Cytosine. However, many alternate H-bond pairs can be formed, with resulting structures that are stable but not double-helices.
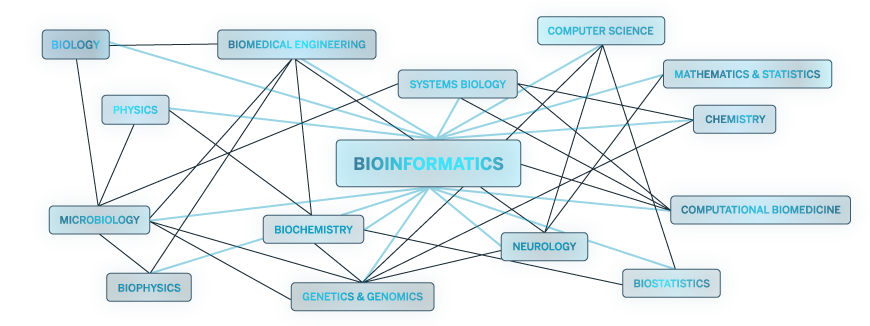
- Bioinformatics an interdisciplinary field which examines biological problems using computational methods, including data collection and organization, file structuring and formatting and storage, analysis via statistical and algorithmic approaches, and specialized summarization and visualization methods. Computer programming and quantitative analysis skills are required, as well as a deep understanding of biological processes and techniques.
- Biology- the study of life, or living matter, in all its forms and the full range of phenomena that living systems exhibit under external conditions. Particular focus areas are often divided into origins of life (also, synthetic biology), cell/organismal growth, cell differentiation and development, reproduction, molecular/system/cell/tissue/morphological/ population/ecosystem structure, and behavior.
- BLAST- acronym derived from Basic Local Alignment Search Tool. An algorithm using a seed sequence and building outwards with a scoring function to find limits, this method finds regions of local similarity between sequences. These sequences can be DNA, RNA, or amino acid, but only one. Software programs have been written to make the algorithm easy to use reference sequences are often pulled from databases of many sequences, and the scoring function usually has parameters that a user can select to decide how imprecise a match is still of interest.
C:
- Chloroplast organelles found in plant cells and eukaryotic algae that absorb sunlight and use the energy of the photons to convert carbon dioxide and water into sugars, the primary source of energy for the cells. They have their own chromosome, which replicates without recombination and independently of nuclear chromosomes. The chloroplast genome size varies, the length of the tomato chloroplast (for example) is ~160,000 nt.
- Computer Science-the scientific discipline that deals with designing hardware to carry out computation, as well as the computational theory for using that hardware, and the development of applications, as software methods, for processing files of input data to efficiently provide output results that closely map to the expected theoretical results.
- Consensus sequence the most commonly occurring amino acid or nucleotide at each position on an aligned series of biomolecules (protein or nucleic acid).
- Conserved sequence: A sequence within DNA or protein that is consistent (shows few changes) across species over a long evolutionary period.
- Contig a series of sequences that are ordered such that there is sequence overlap of each member to two others (except on the ends). Thus there is redundancy at many of the positions, because multiple reads give the same sequence information, but the effective sequence is much longer than any one component.
D:
- Database- a comprehensive collection of data, stored in a digital format, housed in a software system of applications that allow for convenient access and retrieval. Data sets are related in some fashion that is meaningful to a specific purpose for example an organism database may house all molecular, behavioral and morphological data that has been measured and collected about the organism. Another database might have all of the DNA sequences of particular genes from every organism for which sequence has been obtained. The two databases might be logically linked by having DNA sequence data about the same organism.
- Distance matrix: The method used to present the results of the calculation of an optimal pairwise alignment score. The matrix field (i row length for the sequence of one biomolecule in the pair, j column length for the sequence of the other biomolecule in the pair) is the score where the row and column intersects, and the value is assigned to give the optimal alignment between two residues from the input sequences.
- Distance measure: A function is used to associate a non-negative numeric value with a comparison of two biosequences. Similar sequences will have a short distance between them in the function space. Conserved sequences will have a score reflecting a short distance, divergent sequences will have a score reflecting a large distance. There are many types of distance measures, all are based on some assumptions about how sequences change over time and what this means. A distance measure can also be developed for the shape of biomolecules, or their functions.
- DNA- the acronym of Deoxyribo-Nucleic Acid. Ribose is the common sugar in all DNA and RNA monomers, in this case one hydroxyl (-OH) group that is present in RNA molecules is replaced by a hydrogen (-H) atom, thus de-oxy-ribose, the common sugar in DNA molecules.
- DNA Bar Code a technique for characterizing species using a short DNA sequence that is characteristic of the species but varies between species. Depending on the level of discrimination you want, different sequences may be used; microsatellite sequences are commonly used for identifying individuals within a population, for example, but may not be useful for comparing different species.
- DNA Subway a computational resource whose entry point is hosted by Cold Spring Harbor Laboratories and supported by resources from CyVerse. A collection of tutorials is provided as well as means for accessing sequence analysis programs via Web interfaces. The programs are organized as sets of pipelines, so that a task requiring multiple steps can be properly connected together and used efficiently.
- Dot Plot- In comparing two sequences, there are many ways to visualize how the subsets of a each sequence that are similar and those that are different are arranged. The dot plot is one such, very simple, visualization tool.
E:
- E-value: a parameter in the BLAST suite of programs that describes the number of hits, that is sequences for which match = yes given your criteria, that you would expect to see by change when searching a database of the particular size you have referenced. The exponent will be a large negative value when the chance of a random match is very small. The quality of this match will depend on the length of the alignment (you are more likely to pick up shorter sequences by chance) and the degree of similarity ( you are more likely to get a match with a short sequence where you have allowed a high percentage of mismatches to still be qualified as match = yes).
- EMBL Nucleotide Sequence Database: Europe's primary nucleotide sequence resource, which mirrors the sequence data in GenBank, the US repository maintained by NCBI. Individual scientists may submit sequence data, and large genome sequencing centers also submit data The database is produced in collaboration with NCBIs GenBank (USA) and the DNA Database of Japan (DDBJ). Each of the three groups collects a portion of the total sequence data reported worldwide, and all new and updated database entries are exchanged between the groups on a daily basis.
- Entrez: The user interface to a set of Web-accessible databases that store, organize and allow the retrieval of biosequence data, and related data about expression, phenotype, genetics, etc.. The series of programs that enable the resource was created by the National Center for Biotechnology Information (NCBI), a division of the NIH.
F:
- FASTA: An alignment algorithm, for which a number of software programs have been written. It is also used to refer to the data format required to input sequence to the early implementations, which became a standard. The program is one of the many heuristic algorithms proposed to speed up sequence comparisons, by prescreening sequences to locate the highly matching segments between two sequences first, and then extending these matching segments to create local alignments. The sliding window approach that looks at all possible comparisons is more complete but very slow when the sequences are long (for example the Smith-Waterman algorithm).
- Fingerprinting: using a set of DNA fragments that contain sufficient variation that the combination of sequenced fragments will identify and individual to a very high level of certainty. Used in DNA forensics, for example, although there are many other applications.
- Functional genomics: The development and application of experimental and computational approaches to assess all of the gene functions present in the products of the set of genes expressed in a sample at a particular point in time under particular conditions. Genomics, transcriptomics, proteomics, metabolomics and structural genomics data are integrated to create a functional genomics profile.
G:
- Gap- when aligning two sequences it may become clear that by introducing a gap the sequences line up much better. This gap can be introduced generally a penalty is enforced since the sequences under comparison are different (otherwise it can be hard to score missing data).
- Gene-the basic physical unit of heredity; a linear sequence of nucleotides along a segment of DNA that provides the coded instructions for synthesis of RNA. Some of the end products are RNA molecules and some are proteins. Cellular functions are carried out by the RNA and proteins, sometimes being structural and therefore contributing directly to the final phenotype of the organism, and sometimes they are regulatory or enzymatic, meaning there may be additional steps before the end phenotype occurs (such as enzymes that process sugar molecules to make starch).
- GenBank: The NIH genetic sequence database, which includes all sequences (DNA, RNA and protein) both measured experimentally and inferred computationally, with labels indicating informative sub-sequences that have important functional and regulatory roles. The repository has a Web interface at http://www.ncbi.nlm.nih.gov/.
- Genetic polymorphism: The occurrence of one or more different alleles at the same locus in a one percent or greater of a specific population.
- Genome- all the inheritable traits of an organism, including chromosomes, mitochondria, chloroplasts, plasmids and similar replicating units.
- Genomics- the technologies and computational tools used to acquire and assemble all of the genome sequence of an organism. Also used to indicate the study of complete genomes and their structure, rather than individual genes.
- Genotyping: The use of markers to organize the genetic information found in individual DNA samples and to measure the variation between such samples.
H:
- Heuristics- in computer science this refers to a technique designed to solve a problem quickly, using some approximation, when classic methods are too slow, or an exact solution may not be possible (such as NP hard problems). The method trades off optimality, completeness, accuracy or precision, depending on what is considered most important in the application of the results the result is good enough even though it might not be best.
- Hidden Markov models (HMM): A model that a number of algorithms implement as computer programs, the model is built so that it locates the essential, unique features which distinguish (for example) a protein or gene family by analyzing a range of known sequences from the family and determining the combination of features that give the best discrimination. These features then are used to locate similar characteristics in unknown sequences.
- Homology- in the context of Biology, homology is the existence of shared ancestry between a pair of genes, gene products or structure in different taxa. For structures, you could compare a bats wing and insect wings, which functionally do the same thing for motion, but did not arise from common descent, compared to the wings of bats and the arms of primates, which did. Or you could consider photoreceptors in eyes, which have evolved independently a number of times a sequence comparison will show that in some cases there is homology while for others there is not.
- Homology modeling: The use of 3-dimensional (3-D) geometry and sequence information from proteins of known 3-D structure to develop models for proteins whose 3-D structure is unknown. In the first step of homology modeling, search and alignment algorithms are used to find the best sequence overlap of the 'unknown' protein with the sequences of related proteins which have 3-D data. In the second step, the geometry of the 3-D structures is used as a template for generating a 3-D structural model for the regions of high sequence homology in the unknown protein (the conserved regions). Finally, the sections with low homology to known proteins (the variable regions) are modeled using a variety of computational techniques.
I:
- Inversion- part of a sequence may be incorrectly inserted into a chromosome in a back-to-front manner in a coding sequence this will usually result in a mutation of the gene, in a regulatory region this may cause other mistakes in response.
J:
- JASPAR- a database of sequences that code for a particular type of gene called a transcription factor, a gene that binds upstream of other genes and affects when they are turned on and off and how much.
K:
- Kilobase (kb): A length of DNA equal to 1,000 nucleotides.
- K-mers all the substrings of length k that you can find in a given sequence. In computational genomics, k-mers are all the possible subsequences of the selected length in a sequence read. If the sequence has length L, then there will be L-(k+1) possible substrings. The number of possible k-mers is nk for DNA since there are 4 nucleotides.
L:
- Loci: The location of a gene or other marker on the surface of a chromosome. The use of locus is sometimes restricted to mean regions of DNA that are expressed.
M:
- Mismatch Limit-a parameter that can be set in algorithms that measure sequence similarity - determines how similar the two sequences in a window must be to match. For example, if the comparison length (window size) is 9 and the mismatch limit is 2, then only zero to two mismatches will lead to a match=yes score.
- Mitochondria an organelle found in large numbers of eukaryotic cells this is where the biochemical reactions of respiration (converting oxygen and nutrients to ATP for energy production) are located. Mitochondria have their own circular genome, that replicates independently of the nuclear chromosomes and does not undergo recombination. The genome size varies tremendously depending on the organism, from 16, 569nt for humans to 570,000nt in maize.
- Motifs: A pattern of DNA sequence that is similar for genes of similar function. Also a pattern for protein primary structure (sequence motifs) and tertiary structure that is the same across proteins of similar families.
- Mutations- a change in the sequence of a polymer (DNA, RNA or protein). The mutation can be a single base substitution, a gap, and insertion, a very large inversion etc. Some mutations are considered silent if the change in DNA causes the same amino acid to be used because of the redundancy of the triplet code the change is often labeled as silent, although in fact phenotypic changes are known to occur in response to some silent mutations.
N:
- Next generation sequencing -also known as high-throughput sequencing, is the term used to describe a number of different modern sequencing technologies that use sequencing by synthesis but do not use traditional Sanger di-deoxy chemistry, and have a very high parallel sequancing capacity. The length of an individual sequence read is somewhat less than the older Sanger/fluorescent dye chemistry could achieve. The two primary platforms are from Illumina and Thermo-Fisher/Ion torrent. A third-generation method, from Pac Biosciences uses single-molecule sequencing in a a nanopore membrane to acquire very long reads but a much lower level of parallel sequencing and at a higher error rate.
P:
- Pairwise alignment: A comparison of two sequences of the same biomolecular type. In the first step, two sequences are padded by gaps so that they are the same length and so that they display the maximum similarity on a residue to residue basis. An optimal Pairwise Alignment is an alignment which has the maximum amount of similarity with the minimum number of residue 'substitutions'.
- PDB (Protein Data Bank): An international repository for the results of macromolecular studies using NMR, X-ray crystallography, or homology methods. The results of structural studies of proteins, RNA, DNA, viruses, and polysaccharides are presently available. The term PDB also defines a standard file format for publishing protein and nucleotide structures for use in computer programs.
- Personalized Medicine- a type of medicine where a doctor has the entire genome of the patients, and through analysis of the genome, predictions about the possible diseases, predispositions, mutations, or disorders an individual might have. In particular, how receptors and enzymes interact with small molecules (most drugs being small molecules) may show that at individual will respond well to a therapy, may not respond, or may have a negative response.
- Polymorphic marker: A length of DNA that displays population-based variability so that its inheritance can be followed.
- Polymorphism: Individual differences in DNA. Single nucleotide polymorphism (the difference of one nucleotide in a DNA strand) is currently of interest to a number of companies.
- Proteomics- the technical processes and computational methods used to identify all proteins that are present in a sample under given conditions. This is generally the parts list while structural bioinformatics handles what we can understand about how the molecules are folded in space and how this affects their functional capabilities.
- Pipeline designed to accomplish a similar task repeatedly and reproducibly, a pipeline is a set of programs that pass their output, properly formatted, and invoking the next program required in the series. Sometimes referred to a workflow management systems. Galaxy is a popular environment for setting up and running genomics data analysis pipelines. DNA Subway is a similar environment whose purpose is to train scientists in the use of such pipelines.
- Phylogeny an inferred evolutionary relationship, obtained by comparing and scoring characters (such as DNA sequences, or development pathways, or morphological characteristics) between organisms. The comparison of similarities leads to a hypothesis as to where the ancestors of organisms branched away from one another. A classical way to represent this structure is with a phylogenetic tree, but other visualization modes are known.
Q:
- Quality Score- sequencing quality scores are a measure of the probability that a base has been correctly identified (or called). The quality score of a given base Q, is defined by the equation Q = -10log(e) where e is the estimated probability (from error models and prior information about the chemistry and detection methods) that the base was incorrectly identified. A higher Q-score means a smaller chance of error. Q10 means there is 1 change in 10 of an incorrect call, so the inferred accuracy would be 09%, while Q30 means 1 error in 1000, or a 99.9% chance of accuracy. Different sequence platform providers have different error models and determine the Q-score in different ways, so a Q30 from Illumina does not necessarily correspond to a Q30 from Ion Torrent.
R:
- Regulatory region: The segment of DNA that controls whether and to what degree, a gene will be expressed.
- RNA- the acronyms for Ribo-Nucleic Acid. The ribose sugar is common to all 4 of the nitrogenous bases (ACGU). RNA is the material transcribed (copied) from DNA, when it codes for proteins it is called messenger RNA (mRNA), which interacts with tRNA on the ribosome to produce proteins. There are many RNA molecules that carry out cellular functions (often regulatory functions) in the absence of translation to protein, in addition to the ribosomal RNAs (rRNAs) and tRNAs.
S:
- Scaffold: A series of contigs that are in the correct order, but are not connected in one continuous length.
- Scoring Function (also cost function or weight function): The methods used to evaluate the quality of the overlap between sequences. A variety of scoring functions are used to evaluate single replacement operations, multiple alignments (either whole or columns), and pairwise alignments. The score of an alignment of two sequences (a and b) is the sum of the score of all the replacement operations that lead from a to b.
- Sequence- the linear arrangement of monomers belonging to a polymer, whether nucleotides or amino acids. This order generally corresponds to a piece of biological information (for example the sequence of a protein shows the backbone connection of the amino acids) although one sequence may include subsequences that are combined in different ways to create specialized products.
- Sequencer an instrument that, combined with a particular chemistry, allows the linear arrangement of monomers in DNA to be assayed. The chemistry used produces a signal that the sequencer detects, each signal is associated with a particular monomer and time which allows inferring the order of nucleotides in the sample. Signal detection software compares the signal to an error model to provide a base call (which base is present) and a quality score (how likely the base call is to be correct). There are protein sequencers (although mass spectrometry is more often used for proteins) but the assumption is that a DNA sequencer is meant when sequencer is used.
- Similarity measure (also similarity function or similarity score): The result of a scoring function that is used to rank the degree of similarity of a pair of sequences. Larger values indicate greater similarity.
- Single nucleotide polymorphism (SNP): The most common type of DNA sequence variation. An SNP is a change in a single base pair at a particular position along the DNA strand. When an SNP occurs, the gene's function may change, as seen in the development of bacterial resistance to antibiotics or of cancer in humans.
- Structural genomics: The prediction of the 3-D structure of proteins encoded by genes using both experimental and computational techniques.
Substitution Matrix. described the rate at which one character in a sequence changes to other character states over time. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments where the similarity between sequences depends on their divergence time and the substitution rates as represented in the matrix.
- Synthetic Biology the convergence of biology, including biotechnology, computational biology, molecular biology, systems biology, biophysics, computer engineering and genetic engineering seeks to use engineering principles to design biological components with well-defined properties that result in predictable outcomes, as biological machines, when combined. The components do not have to derive from pre-existing biological systems, for example there is work on using a 4-nucleotide codon recognition system, or tRNAs for amino acids that are not naturally occurring.\
- Systems Biology- systems biology is the study of networks of biological components and how they interact, regulate, modulate and function as a whole. The components may be molecules in a cell, cells in a ecological setting or in a tissue, more complex organisms, or populations. Such systems change over time (they are dynamic) and very complex, with internal feedback and feedforward loops and interactions. Thus the behavior of the network often cannot be predicted based on studies of individual components. This has required advanced technologies for gathering and organizing data across the system simultaneously, and the development of new analytical procedures.
T:
- Threading: Computational algorithms which use the known 3-D structure of proteins as a template for positioning (or 'threading') an unknown sequence. The overlap of the sequences is based on how the sequences match with respect to physical properties at specific residues rather than matching based on the similarity of amino acid sequences.
- Transcription: The process of copying a strand of DNA to yield a complementary strand of RNA
- Transcription Factors: The class of proteins which bind to DNA and promote or inhibit the initiation of transcription.
- Transcriptomics: the technologies for acquiring the complete set of RNA sequences present in a sample, and the computational tools used to quantify and compare that set to other sets obtained from samples obtained under different conditions or related samples. For example, if yeast are grown with and without a specific nutrient, a comparison of the transcription profiles will show which genes are specifically turned on (or off) in response to the presence of the nutrient.
W:
- Window size- a parameter that is set when an algorithm is used to compare biological polymer sequences. This tells the algorithm how many contiguous monomers are to be included in a particular comparison.
|
Background
|
Web-accessible papers giving more in-depth background about the topics and tools
DNA Subway:
BLAST
A series of pages from Julians Science Experiments, with good descriptions and some experiments you can try.
|
Home
|
Web Links to Tools
|
Files
|
Schedule
|
|
|